|
|
In the neurological evaluation of weakness, we distinguish between upper
motor neuron weakness, and lower motor neuron weakness. The differences
are tabulated below.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fasciculations are irregular contractions of a group of muscle fibers innervated by one axon. Clinically this appears as a small muscle twitch.
It is also customary, and very helpful, to classify LMN weakness on the basis of the anatomical station affected.
These stations are:
1. The anterior (ventral) horn cell
2. The peripheral nerve, (ventral and dorsal
nerve roots i.e., radiculopathy or nerve i.e., neuropathy)
3. The neuromuscular junction
4. The muscle (i.e. myopathy)
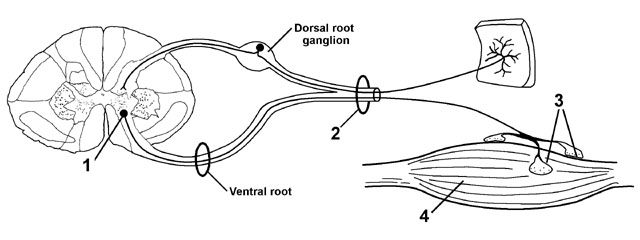
Figure 1 The
4 anatomic stations underlying lower motor neuron weakness
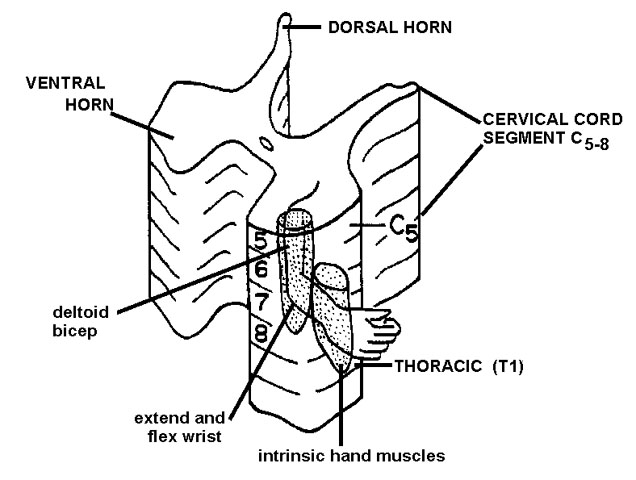
The anterior horn cells are somatotopically organized in the spinal cord. That is, medially located anterior horn cells innervate the proximal muscles, while laterally located ventral horn cells innervate more distal muscles. The arrangement at cervical segments is shown in figure 2. This organization means that diseases that destroy anterior horn cells can result in highly selective weakness. Not only may a single muscle become weak, but only portions of the muscle may be affected. As a rule however the adjacent anterior horn cells will also be affected with weakness of adjacent muscles.
Nerves
A note on the classification of dorsal and ventral root fibers.
The axons in the dorsal roots have been classified based upon their conduction velocities and their sizes. This has led to some confusion in the literature (and for medical students!!). The classifications scheme based upon fiber size uses Roman numerals. Thus, there are I, II, III and IV fiber types. You already have heard about the Ia fibers and that they are associated with muscle spindles and are large and fast conducting. You also have heard that the Ib fibers are associated with the Golgi tendon organs and are little smaller and slower conducting than the Ias. Also remember that II fibers are associated with muscle spindles but are slower conducting and smaller that the Ias and Ibs. II fibers are also associated with receptors carrying information from encapsulated endings used in two point discrimination, vibration and conscious proprioception. III fibers are smaller than Is and IIs and are only lightly myelinated and relatively slow conducting. Such fibers are associated with cooling and first pain. Finally, IV fibers are unmyelinated and convey second pain and warming.
Now lets turn to the classification that uses letters versus Roman numerals. The largest and fastest conducting fibers are called A fibers. Aa (alpha) fibers are comparable to the Ias and Ibs. Ab (alpha-beta) fibers are equivalent to II fibers in size and conduction velocities. Ad (deltas) are equivalent to IIIs and associated with cooling and first pain B fibers are smaller than A fibers, are lightly myelinated and are visceral afferents; they have no equivalent in the Roman numeral system. Finally, C fibers are unmyelinated and equivalent to IV fibers. In addition to carrying second pain and warming such fibers are postganglionic autonomics (but these do not travel in the dorsal roots).
What about ventral root fibers. The processes of lower motor neurons that innervate extrafusal muscle fibers are Aas (or just alpha motor neurons). The preganglionic autonomic axons in the ventral root are B fibers. Finally, there are axons in the ventral roots that innervate the intrafusal (not extrafusal) fibers of the muscle spindles. These are called Ag (gamma) motor neurons (no equivalent in Roman numerals).
Remember, A and B fibers are myelinated and Cs are not. In
the Roman numeral system, just remember that only the IVs are not myelinated.
This is important, since demyelinating diseases would affect the somatic
and visceral afferents and efferent fibers in peripheral nerves, pain and
temperature would not be affected.
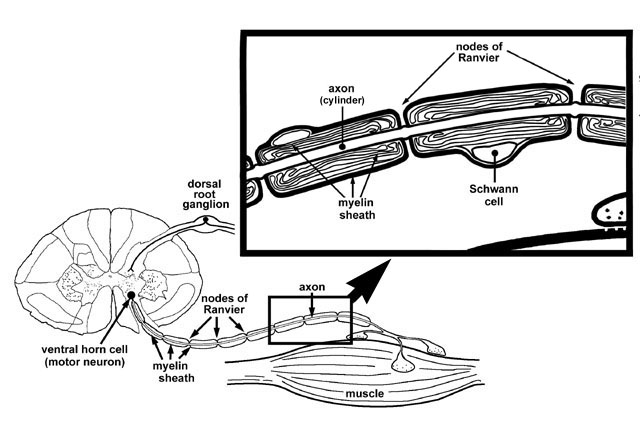
Figure 3 A myelinated nerve fiber
Muscle
One anterior or ventral horn cell, and thus one axon, innervates a few hundred or even a few thousand muscle fibers. The muscle fibers innervated by a single anterior horn cell are collectively known as a motor unit. The "territory" of such a motor unit spans 10 - 15 mm in a muscle, however it is rare that directly adjacent muscle fibers are innervated by the same anterior horn cell / axon. The figure below shows the seemingly random pattern of innervation of adjacent muscle fibers by individual anterior horn cells. The clear fibers below are innervated by a single anterior horn cell and comprise a motor unit. The vertically oriented fibers are innervated by a different anterior horn cell constituting a second motor unit and the horizontally oriented yet another.
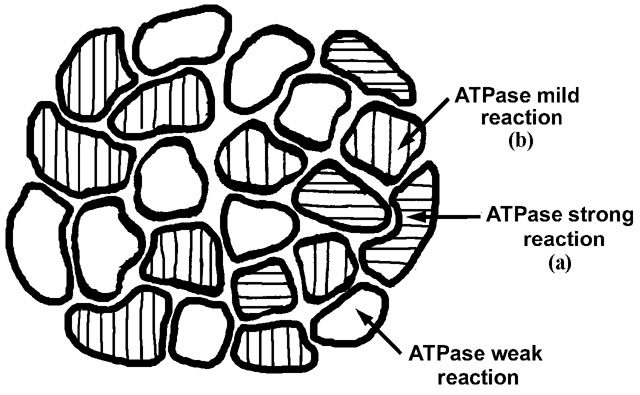
We also need to distinguish between type 1 (slow contracting) muscle fibers and type 2 (fast contracting) muscle fibers. The type of muscle fiber is dependent on the type of anterior horn cell that innervates it. Thus if a muscle fiber is innervated by a type 1 anterior horn cell, it will contract slowly. Certain histochemical reactions, amongst others myosin ATPase, distinguish between type 1 and type 2 fibers. Thus muscle reacted with myosin ATPase will normally exhibit a checkerboard pattern as it is likely that the adjacent muscle fibers are innervated by another anterior horn cell of a different fiber type (figure 4).
Neuromuscular junction
A muscle fiber is activated via a nerve impulse generated by an anterior horn cell. The impulse is conducted along the nerve fiber via saltatory conduction; that is an action potential is generated at one node of Ranvier and then jumps to the next node of Ranvier where another action potential is generated. Once the impulse reaches the neuromuscular junction, voltage sensitive Ca2+ channels are opened which allow for the influx of Ca2+ into the nerve terminal. Ca2+ entry into the nerve terminal initiates the fusion of acetylcholine containing vesicles with the presynaptic membrane and the subsequent release of acetylcholine into the synaptic cleft. Acetylcholine binds to post-synaptic acetylcholine receptors on the muscle membrane. This induces an end plate potential which subsequently results in the generation of an action potential in the muscle fiber membrane (figure 5). The end result of this reaction is muscle fiber contraction.
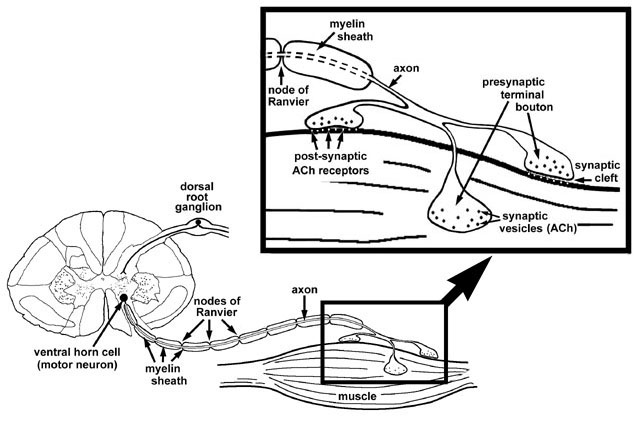
The diagnosis of a specific lower motor neuron syndrome starts with the localization of the disease to one of the 4 anatomic sites. This can be accomplished by a combination of the following investigations:
1) History and clinical examination
In recording the history it is of particular importance to document the following. The time of disease onset, the presence or absence of a family history of other similarly affected individuals, consanguinity (patients born from parent related by blood), the pattern and progression of muscle weakness, the presence or absence of sensory symptoms and the presence of fatigability. The clinical examination serves to corroborate the clinical history, and to document the patterns of weakness, sensory loss, fatigability and reflex changes.
2) Histological examination of muscle or nerve biopsy specimens
These will be dealt with in more detail during the neuropathology section of you Pathology course.
Muscle histology
Muscle is not too smart and can only react in a limited number of ways to insult. Thus most primary muscle diseases have non-specific features in common, such as muscle fiber necrosis, evidence for muscle fiber regeneration, structural abnormalities such as centrally located muscle fiber nuclei and an increase in muscle connective tissue (figure 6). Some primary muscle diseases do show diagnostic changes such as nemaline rod formations or central cores. Inflammation in muscle is important as it may indicate a treatable disease.
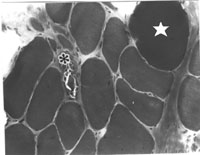
Figure 6 Typical, non-specific pathological findings in a primary myopathy. A necrotic fiber (asterix), and a hypercontracted muscle fiber (star), are shown. The entire muscle is shortened and thus, the hypercontracted fiber is thicker. The connective tissue between the muscle fibers is increased.
Muscle denervation
Anterior horn cell disease or a peripheral neuropathy result in exactly the same histological findings in the muscle! The poor muscle can only interpret these events as "I am denervated." The pathological hallmarks of denervation are type grouping and group atrophy (figure 7). Because one anterior horn cell/motor axon innervates a number of muscle fibers, it follows that disease of an anterior horn cell or its axon results in denervation of a number of muscle fibers. These muscle fibers that have lost their innervation may now be innervated by healthy axons that normally innervate adjacent muscle fibers. The end result is that now one axon innervates more muscle fibers than normal, (a giant motor unit) and also the normal checkerboard pattern of innervation is lost. That is, a whole group of type 1 or 2 fibers can now be seen adjacent to one another (type grouping). With progression of the disease, the axon that sprouted to innervate previously denervated muscle fibers may now also become diseased, resulting in an entire group of adjacent muscle fibers becoming atrophic (group atrophy).
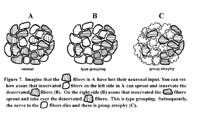
The nerve is equally unimaginative in its reaction to damage. In principle, only two pathological changes are seen. Firstly axonal damage results in Wallerian degeneration, a bead-like disruption of the peripheral nerve that involves both the axon cylinder and the surrounding myelin (Figure 8). This is seen in diseases affecting the axons in the peripheral nerve, or in anterior horn cell disease.

Secondly demyelination results in peripheral nerves with shortened internodes or internodes with thinner myelin (figure 9). Remember the axon cylinder in demyelinating diseases is fine and healthy.

3) Electromyographic (EMG) examination
This test consists of two parts: Nerve conduction studies and needle examination.
a) Nerve conduction studies
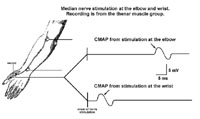
Since there are few pure motor nerves to study, motor nerve conduction recording electrodes are placed over a distal muscle (i.e. thenar muscle group). The appropriate nerve is then stimulated electrically and the evoked responses can be measured. These evoked responses recorded from the surface of the muscle are called a compound muscle action potential (CMAP). The time it takes from stimulation to generation of the CMAP is the conduction speed.
The CMAP represents the action potentials of all muscle fibers activated by the nerve stimulation and the measured response can be compared to a known standard for such stimulation. Reduction in the strength of this response indicates a loss in overall muscle mass or the loss of motor fibers and must further be investigated as to its cause.
For sensory nerve conduction studies, the recording electrodes are placed over superficial nerves (e.g. the sural nerve is a pure sensory nerve). Stimulation of a sensory nerve leads to action potentials in all of the fibers of that nerve and an electrode on the surface of such a nerve records the sensory nerve action potential (SNAP). Furthermore, by stimulating the same nerve over different segments the distances between stimulation sites can be measured and a conduction velocity for the nerve segment established.
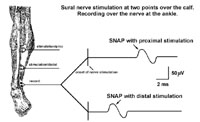
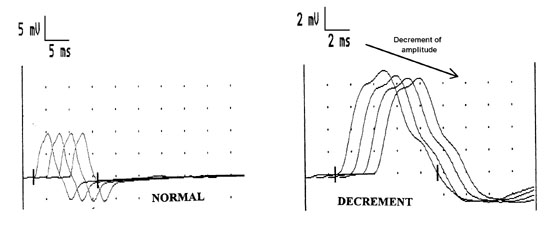
The conduction studies are followed by repetitive nerve stimulation studies. A routine motor nerve conduction study is performed but the nerve is stimulated supramaximally at 2 - 3 Hz and the amplitude of the first 4 CMAPs recorded. In neuromuscular transmission defects the CMAP amplitude decreases with successive stimuli as some muscle fibers are not depolarized due to the neuromuscular transmission defect (figure 12). This is called a decremental response. (The exact mechanism of the decremental response is complex and beyond the scope of this course!! Dont worry!)
Station 1- Anterior (Ventral) Horn Cell disease: This results in low CMAP amplitudes in muscle innervated by the dying anterior horn cells whose axons travel in the nerve being stimulated. There are fewer (than normal) axons that are able to "drive" action potentials in the muscle, the end result being a smaller (than normal) CMAP. Since there is still a population of normal axons from other anterior horn cells (non diseased) nerve conduction velocity is normal, i.e. the nerve (for instance the median nerve) has normal axons that camouflage the dying ones. The sensory nerve conduction studies are normal because ventral horn cells give rise to only motor fibers. Cell bodies of sensory fibers lie in dorsal root or cranial nerve ganglia.
Station 2- Peripheral Nerve disease: The findings will depend on whether both the motor and sensory axons are affected. In most peripheral nerve diseases both become affected. If the changes result in damage only to the axis cylinders the nerve conduction velocities are normal (healthy axons mask the defect), but both the CMAP and SNAP amplitudes will be reduced. If the peripheral nerve disease is predominantly demyelinating (i.e. all of the axons have demyelinated areas) the findings are marked slowing in both the motor and sensory nerve conduction velocities and relatively normal CMAP and SNAP amplitudes (the axis cylinders are OK).
Station 3- Neuromuscular Junction disease: Nerve conduction studies (motor and sensory) are normal, but the hallmark of these diseases is a decremental CMAP response with repetitive nerve stimulation.
Station 4- Muscle disease: Nerve conduction studies are normal, but the CMAP amplitudes will be low, as there is loss of muscle fibers.
In addition to the nerve conduction studies the EMG also involves:
b) The needle examination
An electrode is introduced into the muscle and recordings are made with mild to moderate activation of the muscle. This test is accompanied by some discomfort, but if performed appropriately should not be torture!
Depolarization of muscle fibers in close proximity to the needle electrode will be recorded as motor unit potentials (MUPs; compare with CMAPs!). A normal muscle and a normal MUP are shown in figure 13.
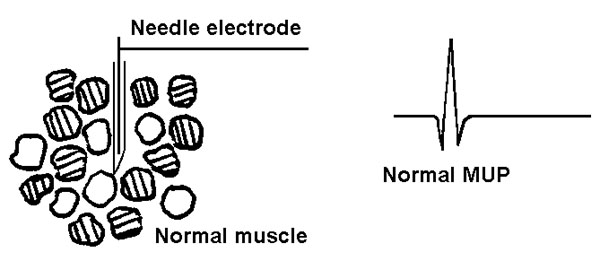
Figure 13 Normal muscle. The above muscle fibers are innervated by three different lower (alpha) motor neurons. Think of the MUP as representing the action potentials of the muscle fibers associated with one of these motor neurons (a motor unit). For example a slight contraction of the muscle during a movement will fire all of the "clear fibers" above, but neither of the "striped fiber" groups.
By analyzing the size of the MUPs (mostly the amplitude and duration), we can make a distinction between diseases that are primarily myopathic (disease of the muscle) versus those which result from denervation. As described in the section on the anatomy of muscle fibers, the muscle fibers innervated by one anterior horn cell / motor axon are spread over 10 - 15 mm of the muscle. Furthermore, the territory innervated by adjacent anterior horn cells overlap so that adjacent muscle fibers are normally innervated by different anterior horn cells or motor axons. With damage to an anterior horn cell or a motor axon the denervated muscle fibers usually become reinnervated by another motor axon with the result that more muscle fibers are innervated by the same anterior horn cell or motor axon in close proximity to the EMG needle. This is seen histologically as type grouping as shown in figure 7. In simplified terms this results in larger MUPs ("neurogenic" MUPs), figure 14. You might wonder why, if there is reinnervation (or "sprouting") and larger MUPs, why are the CMAPs smaller? Well, that is because muscle fibers are also dying (remember group atrophy?)
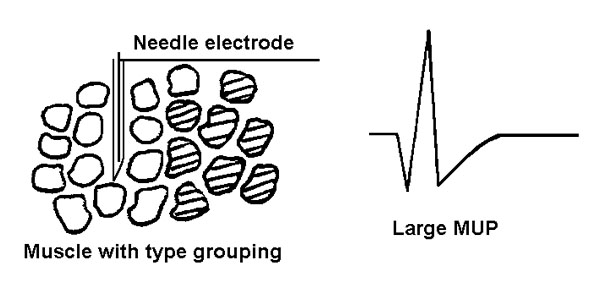
Figure 14 Neurogenic atrophy with type grouping. A large MUP is recorded. Think about the single active motor neuron in Figure 13 as innervating more muscle fibers. When it fires there will be more muscle fiber action potentials and thus a larger MUP.
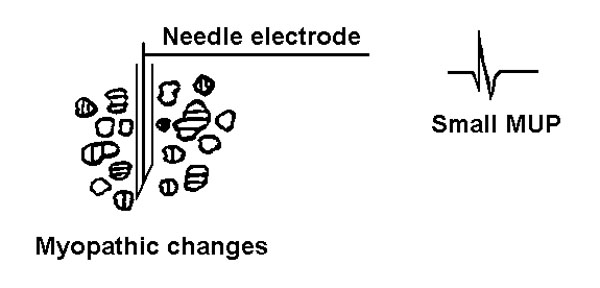
On the other hand in a primary muscle disease, there is loss of muscle fibers, or muscle fibers have a smaller mean diameter than normal, resulting in small MUPs ("myopathic" MUPs), figure 15.
Fasciculations are noted clinically as a contraction of a small group of muscle fibers. They result from the spontaneous discharge of an anterior horn cell or a motor axon with the subsequent contraction of all the muscle fibers innervated by that anterior horn cell or motor axon. Fasciculations can also be recorded with the needle electrode. Clinically, fasciculations are seen after reinnervation of muscle fibers and they are particularly common in amyotrophic lateral sclerosis (motor neuron disease).
4) Biochemical studies
Numerous studies are available but only neuromuscular transmission defects and primary muscle diseases (myopathies) will be discussed.
Neuromuscular transmission defects. In myasthenia gravis, acetylcholine receptor antibodies destroy the post synaptic acetylcholine receptors and they are detectable in blood samples.
Primary muscle diseases - With muscle breakdown of any kind, creatine
phosphokinase (CK) is released into the blood where it can be measured.
5) Genetic studies
The genetic defects of many neuromuscular diseases are now known
and can be detected in peripheral blood or in muscle.
1. Anterior horn cell diseases
Common causes of anterior horn cell diseases are poliomyelitis, motor neuron disease and spinal muscular atrophy. Only spinal muscular atrophy will be discussed further. This is usually an autosomal recessively inherited disease with onset at any time from infancy to adulthood. The primary pathology is the progressive loss of anterior horn cells until the patients become so weak that they die - usually from an associated lung infection. The reason for the progressive loss of anterior horn cells is not clear, but the disease is associated with an abnormality on chromosome 4.
EMG findings: Normal nerve conduction velocities, normal SNAP amplitudes,
low CMAP amplitudes, large MUPs on needle examination, fasciculations.
Histology: Type grouping and group atrophy.
Biochemistry: Defect on chromosome 4.
2. Peripheral nerve diseases
This encompasses a vast number of diseases and only a cursory overview
will be attempted.
Clinical features
Damage to the peripheral nervous system results in motor, sensory and autonomic dysfunction. A neuropathy is any disease of the nerves. There are a number of different classes of neuropathies, but we will consider only one of them here.
Distal polyneuropathy: All the nerves are affected distally in the extremities. Clinically the patients have sensory loss in a glove and stocking distribution, weakness and absent tendon reflexes in distal extremity muscles (e.g. ankle jerk). Longer nerves are affected more severely and thus the changes predominate in the legs. Most distal polyneuropathies are purely sensory or affect the sensory and motor nerves together. Pure motor distal neuropathies are rare. Depending on the etiology, the neuropathies can be axonal (axis cylinder), demyelinating, or show features of both. Diseases that cause distal polyneuropathies include diabetes, toxins, and vitamin deficiency/alcohol abuse. Many of these neuropathies are familial.
EMG findings
a) Predominantly axonal disease: Normal motor and sensory nerve conduction velocities with low or absent CMAP and SNAP amplitudes. Needle examination shows large MUPs that result from denervation and subsequent re-innervation. Fasciculations.
b) Predominantly demyelinating disease: Relatively normal CMAP and SNAP amplitudes with slowed nerve conduction velocities. Needle examination reveals normal MUPs as the axons are not damaged and the muscle fibers are not denervated. In practice pure demyelination is rare and some associated axonal damage is common.
Histology
Type grouping and group atrophy only if there is axonal (axis cylinder)
damage.
3. Neuromuscular transmission defects
Only myasthenia gravis will be discussed further. This disease is characterized by abnormal fatigue with exercise. Myasthenia gravis commonly affects young woman and has a predilection for ocular, facial, masticator and proximal upper extremity muscles. Typically the patients recover to some degree after rest. Thus they feel much better in the morning, but become weaker as the day progresses. When the extraocular eye muscles are affected, diplopia (double vision) and ptosis (drooping of upper eyelid) are common and bothersome signs. This is an auto-immune disease with antibodies destroying the acetylcholine receptors (a postsynaptic defect).
Neuromuscular transmission defects
EMG findings:
Normal nerve conduction velocities, CMAP and SNAP amplitudes.
Decremental response on repetitive nerve stimulation.
Needle examination: Relatively normal MUPs.
Biochemistry:
Acetylcholine receptor antibodies are present in blood.
Histology:
Usually normal.
4. Primary muscle diseases (myopathies)
Muscle dystrophies (dystrophy = faulty development) are genetically determined diseases with onset at any time after birth. They are diagnosed on the pattern of muscle involvement. For example Duchenne muscle dystrophy is characterized by large calves, proximal muscle weakness and weakness of the latissimus dorsi muscles and pectoral muscles. Myotonic dystrophy patients show myotonia (an inability to relax a muscle after contraction) in addition to muscle weakness.
There also are congenital myopathies, metabolic myopathies and inflammatory myopathies that are beyond the scope of this course.
EMG findings:
Normal motor and sensory nerve conduction studies. The CMAPs
are low because of loss in muscle bulk. Needle examination show small
MUPs.
Biochemical findings:
All progressive myopathies have increased CK blood levels indicating
the breakdown of muscle.
Histological findings:
Non-specific myopathic features such as large fibers, necrotic fibers,
and increased connective tissue.
b) Peripheral nerve diseases. Clinically characterized by the associated findings of sensory and autonomic abnormalities. EMG findings depend on whether it is primarily an axonal (axon cylinder) or demyelinating neuropathy; muscle histology shows type grouping and group atrophy with axonal damage; blood CK is normal.
c) Neuromuscular transmission defects. Clinically characterized by abnormal fatigability; EMG shows normal nerve conduction velocities, normal CMAP and SNAP amplitudes, decremental CMAP responses to repetitive nerve stimulation; muscle histology is relatively normal; blood CK is normal.
d) Primary muscle diseases. Clinically specific patterns of muscle weakness
may be noted; EMG shows normal nerve conduction velocities with low CMAP
amplitudes, normal SNAP amplitudes, needle examination shows smaller "myopathic"
MUPs; muscle histology shows myopathic changes; blood CK is elevated.
